
Our science
Convert Pharma’s efforts build on the foundation established by our earlier efforts at Threshold Pharmaceuticals. CP-506 incorporates the best design principles from our earlier efforts at Threshold Pharmaceuticals and Convert’s early results, which include an innovative biomarker, look quite promising. Dr. Barry Selick, former CEO of Threshold Pharma
Tumor hypoxia is known to promote the development of an aggressive phenotype, resistant to chemo, immuno-, and radiotherapy, and is strongly associated with poor clinical outcome. Given its central role in tumor resistance, hypoxia as an exploitable target became an important research area in oncology and the concept of prodrugs, compounds activated by enzymatic reduction in hypoxic tissue, are an attractive solution.
Impact of hypoxia on current treatments
Tumor hypoxia is a driving force for metastasis and resistance to standard of care treatments, including immunotherapy.
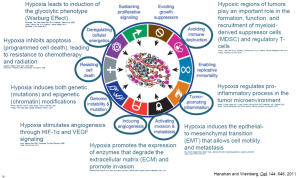
Figure 1: Most of the hallmark of cancer are linked to Hypoxia
Convert has pre-clinical evidence demonstrating that CP-506 has a synergistic anti-tumor effect in combination with radiotherapy, chemotherapy and immunotherapy in the presence of hypoxia.
A potential first to patients hypoxia-activated prodrug
Building on previous science from earlier generations Hypoxia-Activated Prodrugs (HAPs), Convert stands to provide the first hypoxia activated prodrug approved for human use. Convert’s lead compound was rationally designed as a nitroaromatic analogue with highly optimized drug-like properties.

FAQ
What is tumor hypoxia?
Tumor hypoxia is a critical phenomenon in cancer biology. As tumors grow rapidly, they often outgrow their blood supply, leading to areas within the tumor that have inadequate oxygen levels. Hypoxia, thus arising due to a disturbed balance between tumor proliferation and oxygen supply, is a characteristic of ≈50% of solid tumors, e.g. lung, breast, pancreatic and brain cancer. Tumor hypoxia is associated with increased tumor aggressiveness and treatment resistance, representing one of the major bottlenecks in cancer treatment. Hypoxia-related resistances are found in conventional cancer treatments such as chemotherapy and radiotherapy but also in the emerging, vangard cancer treatments such as immunotherapies.
Which cancers present hypoxia?
Hypoxia, thus arising due to a disturbed balance between tumor proliferation and oxygen supply, is a ubiquitous characteristic of solid tumors and present in about 50% of them. Some cancer types have a higher prevalence of hypoxia:
- Glioblastoma: This aggressive brain tumor often exhibits significant hypoxia, contributing to its resistance to therapy.
- Head and Neck Cancers: These tumors frequently develop in hypoxic conditions, which can affect prognosis and treatment response.
- Breast Cancer: Hypoxia in breast tumors can lead to more aggressive disease and poor outcomes.
- Renal Cell Carcinoma: This type of kidney cancer is known to have areas of hypoxia that influence its behavior and treatment resistance.
- Pancreatic Cancer: Hypoxia is a common feature in pancreatic tumors, which can complicate treatment.
- Lung Cancer: Many non-small cell lung cancers (NSCLC) exhibit hypoxic conditions, affecting their biology and response to therapy.
What are the consequences of hypoxia?
Hypoxia has many consequences on cells which are important for cancer treatment, including:
- Hypoxia leads to induction of the glycolytic phenotype (Warburg Effect) (Semenza, Curr Opin Genet Dev 2010; Kroemer, Cancer Cell 2008; Zeng, Cancer Lett 2014; Masson, Cancer Metab 2014; Semenza, J Clin Invest 2013).
- Hypoxia inhibits apoptosis (programmed cell death), leading to resistance to chemotherapy and radiation (Graeber, Nature 1996; Effer, MCB 2004; Weinmann, FASEB J 2004; Koshikawa, Oncogene 2006).
- Hypoxia induces both genetic (mutations) and epigenetic (chromatin) modifications (Bristow, Nat Rev Cancer 2008; Bindra, Cancer Met Rev 2007; Huang, J Mol Med 2007; Bragel, Mediators Inflamm 2010).
- Hypoxia stimulates angiogenesis through HIF-1α and VEGF signaling (Pugh, Nat Rev Cancer 2003; Dehne, Cell Rev Biochem 2009; Wong, Mol Cancer Res 2013).
- Hypoxia promotes the expression of enzymes that degrade the extracellular matrix (ECM) and promote invasion (Pennacchietti, Cancer Cell 2003; Boscolo, Nat Rev Cancer 2010).
- Hypoxia induces the epithelial-to-mesenchymal transition (EMT) that allows cell motility and metastasis (Thiery, Nat Rev Cancer 2003; Tiwari, J Biomed Sci 2012; Vanden, Exp Cell Res 2012; Maniotis, Int J Cancer 2013).
- Hypoxic regions of tumors play an important role in the formation, function, and recruitment of myeloid-derived suppressor cells (MDSC) and regulatory T-cells (Corzo, J Exp Med 2010; Sceneay, Oncoimmunol 2013; Tartour, Cancer Metastasis Rev 2011; Ydotda, Methods Mol Biol 2010).
- Hypoxia regulates pro-inflammatory processes in the tumor microenvironment (Holger, N Engl J Med 2011; Shay, Semin Cell Dev Biol 2012; Maniotis, Int J Cancer 2013; Finger, Cancer Metastasis Rev 2010; Becker, Cancer Immunol Immunother 2013).
- Hypoxia sustains proliferative signaling, allowing for unchecked growth of cancer cells.
- Hypoxia evades growth suppressors, leading to uncontrolled tumor development.
- Hypoxia enables replicative immortality, allowing cancer cells to divide indefinitely.
- Hypoxia promotes tumor-promoting inflammation, enhancing cancer progression and metastasis.
- Hypoxia allows for immune evasion by avoiding immune destruction, protecting tumor cells from the body’s immune system.
Why do some people consider the theoretical concept of HAPs to be one of the holy grails in oncology?
The treatment of hypoxia using Hypoxia-Activated Prodrugs (HAPs) is seen as a “holy grail” in oncology because HAPs target and activate specifically within the low-oxygen regions of tumors, which are typically resistant to conventional therapies. By focusing on these hypoxic areas, HAPs offer a way to finally treat aggressive and hard-to-reach cancer cells while sparing healthy tissue, potentially improving outcomes in otherwise difficult-to-treat cancers. This targeted approach addresses a critical challenge in cancer treatment.
What is CP-506?
CP-506 is a novel, next-generation hypoxia-activated DNA alkylating agent with superior pharmacokinetic properties compared to previous HAPs. We have demonstrated that CP-506 selectively kills hypoxic tumor cellsin vitro and in vivo. In vivo, CP-506 demonstrated potent antitumor effects in a broad range of tumor xenograft models. A causal relationship was established between tumor oxygenation and antitumor effects of CP-506 xenografts as well as significant correlation between baseline hypoxia and tumor response across different tumor types using a multivariate approach.
Why are you excited about what you’re working on?
Tumor hypoxia is a well documented issue for which no solution has been provided so far, leaving patients with sub-optimal outcomes. Being able to work on molecules with such potential is highly motivational.
What impact has been proved in pre-clinical trials?
In vivo, CP-506 monotherapy demonstrated potent antitumor activity, at well tolerated doses, across a broad panel of human tumor xenograft models and significantly inhibited tumor growth in 13 out of the 15 models tested. As we could expect, the 2/15 models where there was no response were in fact not hypoxic.
What is the state of your science and pipeline?
Our main molecule, CP506, is currently going through phase I+IIa clinical trials. We are also acquiring other promising molecules with a synergistic effect with immunotherapeutics, providing a complementary diversification to our portfolio.
Why do you consider the timing for CP506 to be ideal?
An increasing number of people are starting to realize the limitations of standard of care treatments which are hurdled by hypoxia. In the meantime, recent scientific progress for instance on biomarker enable us to better segment patients who are very likely to be good responders to our treatments.
How are you maximizing your chances of success?
Like for the creation of the first airplane or the first polio vaccine, we’re happy to be able to lean on the shoulders of the giants who got close to making this breakthrough yet failed before us. In particular here are the hurdles we cleared:
- HURDLE 1 CLEARED: TOXICITY – PR104, a previous candidate, failed due to myelotoxicity. This was the single serious adverse effect which prevented dose-escalation. We have adapted our formulation and believe we cleared this hurdle.
- HURDLE 2 CLEARED: HIGHER IMPACT DUE TO LOWER METABOLIC LOSS: The di-nitro adopted for PR-104, where the nitro group ortho to the mustard contributes to facile metabolic loss via self-alkylation, is avoided as CP-506 is a mono-nitro HAP.
- HURDLE 3 CLEARED: ORAL BIOAVAILABILITY – CP506 has the potential to be orally bioavailable, unlike previous candidates
- HURDLE 4 CLEARED: BYSTANDER EFFECT: Unlike evofosfamide (TH302), where the released phosphoramidate mustard cytotoxic metabolite is essentially cell entrapped, physiochemical properties of CP-506 and its metabolites readily permit a bystander effect.
Which types of tumors or cancers are targeted by your molecules?
Because all tumors can be hypoxic to some extent, we have a tumor agnostic approach. In the long term we expect even more impact for specific tumors that are particularly hard to treat or especially hypoxic. Our first patient in the clinical trial for instance presents pancreatic cancer.
How confident are you to translate the mice results to the patients?
We’re highly confident for 3 reasons:
- We have learned crucial lessons from previous generations of HAPs, some of which nearly succeeded in Phase III trials, and are advised by their former backers. For instance, CP-506 has been designed to avoid being affected by human aerobic AKR1C3 metabolism, which was a major confounding factor of a previous candidate.
- We have a view on effective dose ranges that are projected to be both therapeutically effective and safe based on preclinical studies and early clinical data.
- Preliminary results from our phase I study appear to confirm this confidence, supporting the translation of the positive effects seen in mice to human patients.
I am a center of research, can we join the study? I’m an investor, is there a way to invest?
Contact us at info@convertpharma.com